Blog Post
Why Depression Studies So Often Fail: Don’t Blame “Placebo Response”
June 29, 2017
 Jack Modell, Vice President and Senior Medical Officer, is a board-certified psychiatrist with over 35 years of experience in clinical research, including 20 years conducting trials, teaching, and providing patient care in academic medicine, and 15 additional years of experience in clinical drug development (proof of concept through market support), medical affairs, successful NDA filings, medical governance, drug safety, compliance, and management within the pharmaceutical and CRO industries. Jack has authored over 50 peer-reviewed publications across numerous medical specialties and has lead several successful development programs in the neurosciences. Jack is a key opinion leader in the neurosciences and is nationally known for leading the first successful development of preventative pharmacotherapy for the depressive episodes of seasonal affective disorder.
Jack Modell, Vice President and Senior Medical Officer, is a board-certified psychiatrist with over 35 years of experience in clinical research, including 20 years conducting trials, teaching, and providing patient care in academic medicine, and 15 additional years of experience in clinical drug development (proof of concept through market support), medical affairs, successful NDA filings, medical governance, drug safety, compliance, and management within the pharmaceutical and CRO industries. Jack has authored over 50 peer-reviewed publications across numerous medical specialties and has lead several successful development programs in the neurosciences. Jack is a key opinion leader in the neurosciences and is nationally known for leading the first successful development of preventative pharmacotherapy for the depressive episodes of seasonal affective disorder.
Prior to joining the pharmaceutical and contract research organization industries, I was in clinical practice for twenty years as a psychiatrist and medical researcher. And something I noticed very early on among my patients with major mental illnesses, particularly those with severe depression and psychotic disorders, was that they did not generally get better – at least not for more than a day or two – by my simply being nice to them, treating them with ineffective medications (e.g., vitamins when no vitamin deficiency existed), seeing them weekly for office visits, or by providing other so-called supportive interventions that did not directly address the underlying illness. To be clear, this is not to say that kindness and supportive therapy are not critical to the patient-physician relationship (“The secret of the care of the patient is in caring for the patient” [Frances Weld Peabody, 1927]), but rather that kindness and support alone rarely make a biologically based illness substantially improve or disappear.
With this background, I vividly recall my surprise upon being asked shortly after I joined the pharmaceutical industry: “Can you help us figure out how to decrease the nearly 50% placebo-response rate we see in antidepressant trials for major depressive disorder?” “Fifty percent?” I replied, incredulously. “There’s no way that 50% of patients in a true major depressive episode get better on placebos or just by seeing the doctor every couple of weeks!” “Seriously?” was the reply, and they showed me voluminous data supporting their figure.
I spent the next few years trying to figure out this apparent paradox. Not surprisingly, the answer turned out to be multifactorial. After careful review of internal and external data, as well as published explanations for high “placebo response rates” in clinical depression trials (much of which also applies to clinical trials in general), the following three factors emerged as being of particular importance because they are easily mitigated by proper trial design, thorough research staff training, and meticulous oversight of study conduct.
(1) Subjects being admitted into clinical trials often had depressive symptoms, but did not truly meet criteria for major depressive disorder. Examples include subjects with personality disorders whose symptoms wax and wane considerably with external factors (e.g., family or job stress), subjects with depressive symptoms in response to a particular stressor (not of sufficient severity or duration to meet formal criteria for a major depressive episode and likely to abate with the passage of time), and subjects who – for various reasons – may feign or exaggerate symptoms for the purpose of seeking attention or gaining access to a clinical trial. Unlike the patients I encountered in my clinical practice, subjects with these presentations often do improve with supportive interventions and placebo.
Recruitment of truly depressed subjects is made even more difficult by the widespread availability of reasonably effective medication options. Patients in the throes of a major depressive disorder, who sometimes have difficulty even making it through the day, are rarely keen to commit to the additional efforts, uncertainties, and treatment delays involved with a clinical trial when an inexpensive prescription for an effective generic antidepressant can now be filled in a matter of minutes. Indeed, as more and more generally safe and effective medications have become approved and readily available for a variety of illnesses, the motivation for patients to join clinical trials in the hope of finding an effective treatment has correspondingly decreased.
(2) The second factor is somewhat difficult to discuss because it sometimes provokes an understandable defensive response in clinical investigators. Consciously or unconsciously, many investigators and clinical raters inflate or deflate clinical ratings to enable the subject to gain entry into, or remain enrolled in, a clinical trial. Most commonly, this is done by subtly – and sometimes not so subtly – coaching subjects on their answers, or when subject responses or findings seem to fall in between scale severity ratings, by rounding up or down to a rating that is more likely to qualify the subject for the trial.
The effect of this practice is diagrammed in the following figures, specific examples of which can be seen in these references.1-3 In Figure 1, the white bell-shaped distribution is the expected distribution in severity rating scores of an unselected clinical population presenting for clinical trial participation, let’s say with a mean score at shown at X̄n. Not uncommonly, what we see in clinical trials in which a certain scale severity score is required for study entry (depicted by the vertical light blue line, with a score to the right of it required for entry) is not the expected right half of this bell-shaped distribution, but rather a distribution like that shown by the orange curve, which is essentially the right-half of the bell-shaped distribution with a large proportion of subjects whose ratings fell short of required severity for study entry (to the left of the blue line) “pushed” to the right, over the blue line, so that the subjects now qualify for study inclusion, with the mean of those thus selected shown at X̄s.
Figure 1
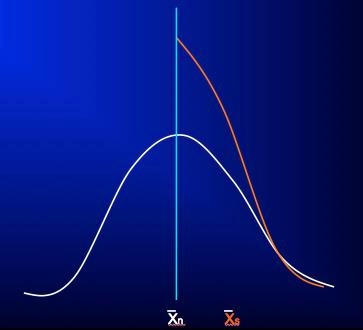
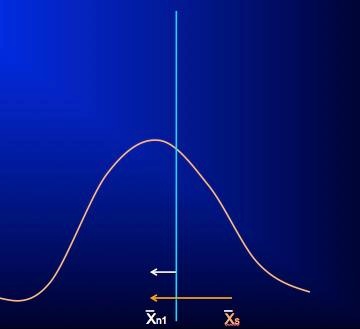
At the first follow-up visit, when raters (and subjects) now have little incentive to influence rating scores to meet a pre-specified criterion, the scores of the entire included population are free to relax towards their true values and assume the pre-selection and more normally distributed pattern. Moreover, subjects and investigators, expecting that the onset of treatment should coincide with at least some clinical improvement, may bias rating scores during this period to reflect this expectation even though the signs and symptoms of the illness may have yet to show true change. During this same time, any actual clinical improvement will also result in the rating score mean shifting leftward (white arrow, figure 2), but because the measured change – from the initial X̄s of the selected population to the new mean (X̄n1; orange arrow, figure 2) – is generally much greater than a true treatment effect during this period, any real changes are obscured and the ability to detect a true drug-placebo difference may be lost. While this early “improvement” in rating scores for subjects in clinical trials may appear to be a “placebo effect” and is often confused with it, this apparent improvement is instead the result of artificially inflated scale scores regressing back to their original true distribution, in combination with whatever actual treatment and placebo effects may have occurred during this time. Unfortunately, the introduction of non-qualified subjects to the study and rater bias will continue to hamper detection of actual drug-placebo differences throughout the course of the study.
Figure 2
(3) Finally, investigators and site staff often do not fully understand the true objective of the clinical trial: it should never, for example, be “to show treatment efficacy” or to show that a product is “safe and well tolerated,” but rather, to test the null hypothesis of no treatment difference or to estimate likely treatment effect, as well as to faithfully and objectively record all adverse effects that may emerge during treatment. Likewise, investigators and site staff often fail to understand the importance of complete objectivity and consistency in performing clinical ratings, the intention behind and importance of every inclusion and exclusion criterion (necessary for their proper interpretation and application), and the destructive effect on the outcome and scientific integrity of the trial that even well-intended efforts to include subjects who are not fully qualified can have.
Each of these three factors can skew both drug and placebo trial populations and results, making it appear that subjects “improved” well beyond what would have resulted had there been strict adherence to protocol requirements and objective assessment of study entry and outcome measures.
What, then, can be done to prevent these problems from sabotaging the results of a clinical trial? Foremost are thorough and meticulous investigator and rater education and training. All too often, perfunctory explanations of the protocol and clinical assessment tools are provided at investigator meetings, and “rater training” takes the form of brief demonstrations of how the rating scales are used and scored, without actually testing raters to be certain that they fully understand how the scales are to be used and interpreted, including understanding scoring conventions, criteria, and necessary decision-making.4 Even seemingly sound training has marked limitations both immediately and as training effects deteriorate during conduct of the trial.4-7
Training of the research staff must include not only a review of the protocol design and study requirements, but detailed explanations about why the trial is designed exactly as it is, the importance of strict adherence to study inclusion and exclusion criteria, and the necessity for complete honesty, objectivity, and consistency in conducting the clinical trial and in performing clinical assessments. Detailed training on the disease under study is also important to ensure that all site staff have a complete understanding of the intended clinical population and disease being studied so that they can assess subjects accordingly. And, as noted above, rater training must include not only education on the background, purpose, characteristics, and instructions for each scale or outcome measure used, but trainers, as well as investigators and raters, should be tested for adequate understanding and proficiency in use of each of these measures.
Meticulous monitoring during the course of the study is also essential to ensure continued understanding of, and compliance with, protocol requirements, as well as accurate and complete documentation of study procedures and outcomes. Study monitors and others involved with trial oversight should review data during the course of the trial for unexpected trends in both safety and efficacy data, and not simply for identification of missing data or isolated datum outliers. Unexpected trends in safety data include adverse event reporting rates at particular sites that are much higher or lower than median reporting rates, and vital signs that are relatively invariant or favor certain values over time. Unexpected trends in efficacy data include changes in closely related outcome measures that are incongruent – for example, objective and subjective ratings of a similar outcome differing considerably in magnitude or direction, that are much larger or smaller at particular sites than those observed at most sites, that occur in relatively fixed increments, and that show unusually similar patterns or values across subjects.
Finally, and perhaps most importantly, is that no matter how well-informed or well-intentioned investigators and raters might be, humans simply cannot match computers in objectivity and consistency, including of the kind needed to make assessments based on subject responses to questions in clinical trials. Unless being programmed to do so, a computer cannot, for example, coach a subject on how to respond, nor would it inflate or deflate ratings based on feelings, expectations, response interpretations, or desired outcomes. A computer faithfully asks the same questions every time, following the same algorithm, and records responses exactly as provided by the subject. Indeed, several studies have shown that computerized assessments of entry criteria and outcome measures in clinical trials – in particular interactive voice response systems (IVRS) and interactive web response systems (IWRS) – provide data of quality and signal-detection ability that meet and often exceed that obtained by human raters.1,3,7,8,9 For these reasons, strong consideration should also be given to using IVR and/or IWR systems for assessing study entry criteria and endpoints that allow such use.
The author acknowledges John H. Greist, MD, for his outstanding research and input regarding these important findings and considerations.
References
- Greist JH, Mundt JC, Kobak K. Factors contributing to failed trials of new agents: can technology prevent some problems. J Clin Psychiatry 2002;63[suppl 2]:8-13.
- Feltner DE, Kobak KA, Crockatt J, Haber H, Kavoussi R, Pande A, Greist JH. Interactive Voice Response (IVR) for Patient Screening of Anxiety in a Clinical Drug Trial. NIMH New Clinical Drug Evaluation Unit, 41st Annual Meeting, 2001, Phoenix, AZ.
- Mundt JC, Greist JH, Jefferson JW, Katzelnick DJ, DeBrota DJ, Chappell PB, Modell JG. Is it easier to find what you are looking for if you think you know what it looks like? J Clinical Psychopharmacol 2007;27:121-125.
- Kobak KA, Brown B, Sharp I, Levy-Mack H, Wells K, Okum F, Williams JBW. Sources of unreliability in depression ratings. J Clin Psychopharmacol 2009;29:82-85.
- Kobak KA, Lipsitz J, Billiams JBW, et. al. Are the effects of rater training sustainable? Results from a multicenter clinical trial. J Clin Psychopharmacol 2007;27:534-535.
- Kobak KA, Kane JM, Thase ME, Nierenberg AA. Why do clinical trials fail. The problem of measurement error in clinical trials: time to test new paradigms? J Clin Psychopharmacol 2007;27:1-5.
- Greist J, Mundt J, Jefferson J, Katzelnick D. Comments on “Why Do Clinical Trials Fail?” The problem of measurement error in clinical trials: time to test new paradigms? J Clin Psychopharmacol 2007;27:535-536.
- Moore HK, Mundt JC, Modell JG, Rodrigues HE, DeBrota DJ, Jefferson JJ, Greist JH. An Examination of 26,168 Hamilton Depression Rating Scale Scores Administered via Interactive Voice Response (IVR) Across 17 Randomized Clinical Trials. J Clin Psychopharmacol 2006;26:321-324.
- http://www.healthtechsys.com/publications/ivrpubs2.html